2019-08-30
MALVA: Genotyping by Mapping-free ALlele Detection of Known VAriants
Publication
Publication
Special Issue: RECOMB-Seq 2019, iScience , Volume 18 p. 20- 27
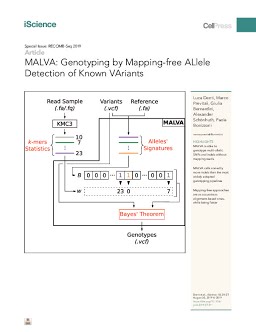
The amount of genetic variation discovered in human populations is growing rapidly leading to challenging computational tasks, such as variant calling. Standard methods for addressing this problem include read mapping, a computationally expensive procedure; thus, mapping-free tools have been proposed in recent years. These tools focus on isolated, biallelic SNPs, providing limited support for multi-allelic SNPs and short insertions and deletions of nucleotides (indels). Here we introduce MALVA, a mapping-free method to genotype an individual from a sample of reads. MALVA is the first mapping-free tool able to genotype multi-allelic SNPs and indels, even in high-density genomic regions, and to effectively handle a huge number of variants. MALVA requires one order of magnitude less time to genotype a donor than alignment-based pipelines, providing similar accuracy. Remarkably, on indels, MALVA provides even better results than the most widely adopted variant discovery tools. Biological Sciences; Genetics; Genomics; Bioinformatics
| Additional Metadata | |
|---|---|
| , , , | |
| doi.org/10.1016/j.isci.2019.07.011 | |
| Special Issue: RECOMB-Seq 2019, iScience | |
| Organisation | Centrum Wiskunde & Informatica, Amsterdam (CWI), The Netherlands |
|
Denti, L., Previtali, M., Bernardini, G., Schönhuth, A., & Bonizzoni, P. (2019). MALVA: Genotyping by Mapping-free ALlele Detection of Known VAriants. iScience, 18, 20–27. doi:10.1016/j.isci.2019.07.011 |
|